
Laboratorio di Chimica Teorica e Computazionale (ThC2-Lab)
![]() Il ThC2-Lab della Sede Secondaria CNR-ICCOM di Pisa coinvolge 4 ricercatori e ricercatrici e numerosi dottorandi ed associati, rappresentando così uno dei più grandi gruppi di ricerca insediati in un unico Istituto CNR e operanti nel campo della modellistica molecolare e della chimica teorica e computazionale.
Il ThC2-Lab della Sede Secondaria CNR-ICCOM di Pisa coinvolge 4 ricercatori e ricercatrici e numerosi dottorandi ed associati, rappresentando così uno dei più grandi gruppi di ricerca insediati in un unico Istituto CNR e operanti nel campo della modellistica molecolare e della chimica teorica e computazionale.
La nostra ricerca è finalizzata allo sviluppo di nuovi modelli, metodi teorici e computazionali, alla loro applicazione allo studio di sistemi e processi rilevanti per la chimica dei materiali e la biologia, e punta ad una descrizione dinamica dei fenomeni oggetto di indagine.
ThC2-Lab ha una consolidata esperienza nello studio delle proprietà ottiche lineari e non lineari di coloranti molecolari in ambienti complessi, ad es. soluzioni di varia tipologia, matrici polimeriche naturali e/o sintetiche, che è stata sfruttata per studiare il comportamento fotochimico e fotobiologico di segmenti di DNA e pigmenti naturali, nonché le proprietà di polimeri funzionalizzati, considerando non solo la loro risposta ottica ma anche la dinamica dei processi innescati dall’assorbimento della luce quali, reazioni chimiche, conversioni interne, energia, trasferimenti di protoni ed elettroni. Con queste premesse si sono recentemente indirizzati parte degli sforzi allo studio di sistemi modello per celle solari sensibilizzate con coloranti e fotosintesi artificiale.
ThC2-Lab ha una consolidata esperienza nella simulazione della struttura, crescita e proprietà di nanocluster metallici e nanoleghe, sia liberi che in ambienti di varia natura come sistemi colloidali e supportati, con applicazioni nella catalisi eterogenea finalizzata a problemi di energia e ambiente, con risultati riconosciuti nella Oxygen Reduction Reaction (ORR) elettrochimica e nella sintesi dell’ammoniaca tramite il processo Haber-Bosch. Anche i materiali a bassa dimensionalità (2D) sia come supporti che per le loro proprietà emergenti nel campo della optoelettronica fanno parte della esperienza del ThC2-Lab. Recentemente, questo interesse si è esteso alla predizione ab initio della morfologia e struttura atomistica di fasi carboniose amorfe e/o nanostrutturate con applicazioni in catalisi, elettrochimica, sensori, e super-condensatori.
Dal punto di vista dei metodi, il personale ThC2-Lab ha esperienza in software dedicati che vanno da DFT, post-HF, QM/MM ibrido, approcci ad onde piane per solidi, modelli di solvatazione impliciti ed espliciti, dinamica molecolare (MD) classica e quantistica. Una parte significativa del nostro lavoro è dedicata allo sviluppo di nuovi modelli, metodi e protocolli computazionali. In questo quadro citiamo metodi per ottimizzare i campi di forza per MD e per simulare spettri elettronici risolti vibronicamente in sistemi grandi e flessibili, metodi di diabatizzazione e frammentazione per valutare accoppiamenti elettronici per trasferimenti di elettroni ed energia, algoritmi di ottimizzazione globale (reattiva) per l’esplorazione di energia potenziale multi-minima superfici tipiche dei nanocluster e delle nanoleghe, e infine metodi di high-throughput screening per il rational design di nuovi catalizzatori e tecniche di Machine Learning. Alcuni di questi sviluppi sono stati implementati in codici open source che sono liberamente condivisi con la comunità scientifica.
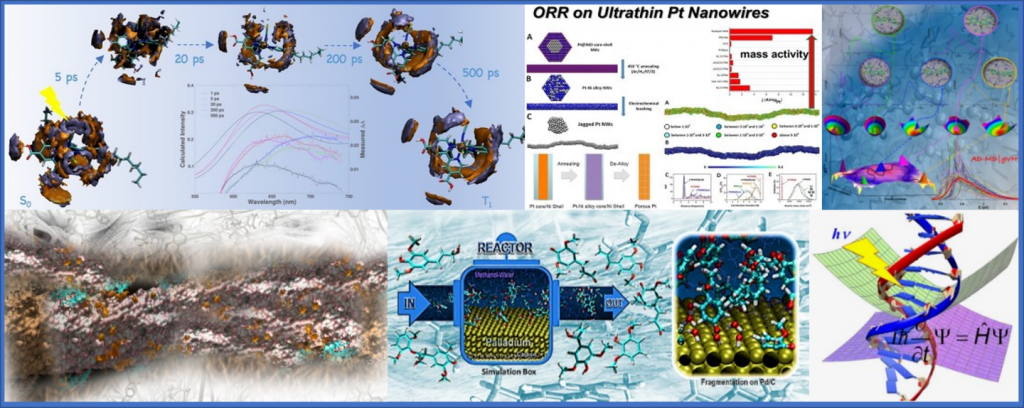
Campi di applicazione
- Sviluppi di modelli e metodi
- Chimica dei materiali
- Energia
- Fotochimica, Fotobiologia e Fotofisica
- Biosensori
- Elettronica molecolare
- Nanocluster metallici e nanoleghe
- Materiali 2D (ossidi, dicalcogenuri, grafene)
- Sistemi di rilevanza biologica
- Spettroscopia computazionale
- Rational Design in Catalisi
- Materiali auto-assemblanti
- Cristalli Liquidi
- Complessi metallo organici per il fotovoltaico
Competenze
- Metodi basati su postHF e DFT per proprietà molecolari in stati elettronici eccitati
- Dinamica molecolare e sviluppo dei campi di forza
- Metodi DFT ad onda piana per solidi e cluster
- Modelli di risoluzione impliciti ed espliciti
- Intersezioni coniche, dinamica quantistica e semiclassica di processi non adiabatici
- Spettroscopia vibrazionale ed elettronica
- Transizioni da stati metastabili
- Progettazione di farmaci
- Metodi multiscala e misti quanto-classici.
- Tecniche di dinamica accelerata e campionamento stocastico
Strumentazioni

- Cluster SoBaSV (440 cores + 30 TB di spazio disco)
- 4 nodi/128 cores CPU Intel® Xeon® Silver 4216 a 2.10 GHz, 128 GB di RAM, 1 TB SATA HD (Vanz1-4)
- 4 nodi/112 cores CPU Intel® Xeon® CPU E5-2680 v4 a 2.40 GHz, 128 GB di RAM, 2 TB SATA HD (Sacco1-4)
- 4 nodi/64 cores CPU Intel® Xeon® CPU E5-2640 v3 a 2.60 GHz, 128 GB di RAM, 2 TB SATA HD (Baleno5-8)
- 4 nodi/64 cores CPU Intel® Xeon® CPU E5-2670 a 2.60 GHz, 128 GB di RAM, 2 TB SATA HD (Baleno1-4)
- 6 nodi/72 core CPU Intel® Xeon® CPU E5650 a 2.67 GHz, 48 GB di RAM, 250 GB SATA HD (Sole)
- Cluster MODULO
- 16 nodi/128 CPU Quad-Core AMD Opteron Processor 2380, 32 GB RAM, 250 GB SATA HD
- Cluster CRESCITA
- 12 nodi/96 CPU Processore AMD Opteron 2354, 16 GB di RAM, 250 GB SATA HD
- 4 nodi / 16 CPU Processore AMD Opteron 265, 8 GB di RAM, 250 GB SATA HD
- DPM cluster
- 8 nodi/64 core CPU Tower HP BladeSystem c3000, 8x HP ProLiant BL460c G1 (2x Intel(R) Xeon(R) CPU E5440 a 2,83 GHz, 16 GB di RAM 150 GB SATA HD)
- FORM e NEWGROWTH clusters
- 16 nodes / 128 CPU Quad-Core AMD Opteron Processor 2380, 32 GB RAM, 250GB SATA HD – Collegamento InfiniBand
- 4 Nodi / 16×2 core – 2 Intel(R) Xeon(R) Gold 6130 CPU @ 2.10GHz – 256 GB RAM – 200GB SATA HD + 4 Nodi / 26×2 core – 2 Intel(R) Xeon(R) Gold 6230R CPU @ 2.10GHz – 192 GB RAM – 100GB SSD + 3 Aree di Storage 3.5 TB SATA HD – Collegamento InfiniBand
Personale strutturato
- Alessandro Fortunelli, Dirigente di Ricerca
- Susanna Monti, Dirigente di Ricerca
- Giacomo Prampolini, Primo Ricercatore
- Fabrizio Santoro, Dirigente di Ricerca